




beta
|
||||||
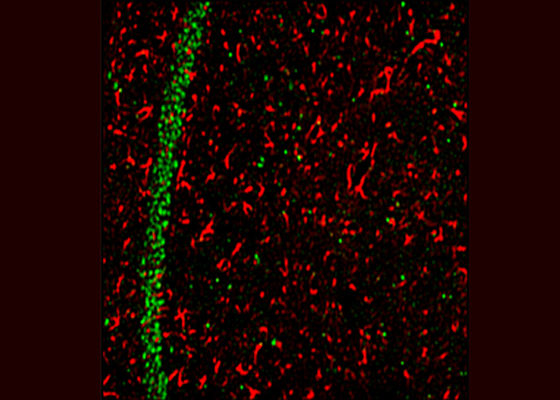 Studies on the Lafora disease mice models
|
Lafora progressive myoclonus epilepsy or Lafora disease (LD) is one of the five forms of inherited progressive myoclonus epilepsies in human and is caused by mutations in the gene coding for the laforin phosphatase or the malin ubiquitin ligase. Besides epilepsy, LD is characterized by the abnormally high level of glycogen and their aggregates as Lafora bodies in many tissues including the neurons. Studies on the Lafora disease mice models - created by targeted deletion of the gene coding for laforin or malin - have demonstrated that in addition to the epilepsy and glycogen accumulation, the animals also show impairment in autophagy – a cellular process where damaged and organelles and protein aggregates are cleared using a vesicle-mediated proteolytic pathway. However the physiological basis of epilepsy LD, and whether the epileptic phenotype is a cause or consequence of glycogen aggregates and/or the autophagic defects have not been established. This study aims to address this very important question by using the mice models of LD (laforin- or malin-deficient mice).
|
|||||



